
A doença de Pompe é uma doença genética neuromuscular progressiva, debilitante, que abrange uma série de fenótipos, variando de um curso rapidamente progressivo, que é geralmente fatal até um ano de idade (“início na infância - IOPD”)1-3, a um curso de evolução mais lenta, mas inexoravelmente progressivo, resultando em morbidade e/ou mortalidade prematura (“início tardio – LOPD”). De acordo com um estudo realizado na Holanda, a incidência da doença de Pompe de início precoce é cerca de 1:138.000, já a de início tardio é de aproximadamente 1:57.000.1-4 No Brasil, estima-se que exista de 1.000 a 3.500 pacientes com a doença.5
Embora considerada rara, a doença de Pompe apresenta um impacto significativo na qualidade e na expectativa de vida. Com sintomas que se confundem com os de outras doenças, é comum que o paciente peregrine por consultórios de diversas especialidades até conseguir um diagnóstico definitivo. Devido à sua rápida progressão, o diagnóstico precoce é essencial para que o paciente receba o cuidado apropriado. Pensando nisso, criamos um conteúdo com informações atualizadas sobre a doença de Pompe, focado em orientar os profissionais de saúde sobre a evolução da enfermidade e as estratégias de manejo, visando encurtar o caminho do paciente até o diagnóstico, a fim de proporcionar a ele uma vida com mais qualidade.
Os músculos do nosso corpo podem armazenar energia por meio de um açúcar chamado glicogênio.
A Doença de Pompe se manifesta quando nosso organismo não produz, ou produz em pequenas quantidades, uma enzima chamada alfa-glicosidase ácida (GAA), responsável justamente por quebrar este glicogênio e produzir energia.3,4,6
Com o tempo, esse açúcar acumulado nas células leva a danos que comprometem a musculatura do paciente, causando fraqueza muscular e fraqueza respiratória.6
Os pacientes são classificados em dois grupos, de acordo com a idade de início: Pompe infantil (IOPD, do inglês infantile-onset Pompe Disease), em que os sintomas aparecem já nos primeiros dias ou durante o primeiro ano de vida; ou Pompe de início tardio (comumente chamada de LOPD, do inglês late-onset Pompe disease), com o início dos sintomas durante a infância (após 12 meses de idade) ou na idade adulta.1,4
Pacientes nascidos com atividade de GAA residual baixa a moderada geralmente tem uma progressão da doença mais heterogênea e menos rápida. Os sintomas começam a qualquer momento da primeira infância até a idade adulta, com pouco ou nenhum comprometimento cardíaco.4
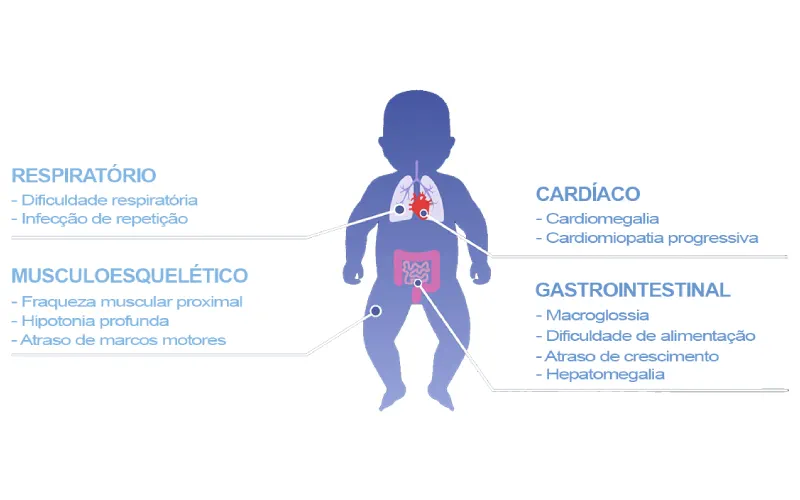
Dentre as manifestações clínicas da doença de Pompe, no IOPD tipicamente se apresenta com cardiomegalia, hipotonia, atraso dos marcos motores, fraqueza muscular, dificuldade de alimentação e dificuldade de crescimento.1,3,4
Adultos e crianças com LOPD apresentam sintomas predominantemente associados à disfunção muscular esquelética, o que resulta em deficiências motoras e respiratórias, com comprometimento progressivo das funções.1,4 A ausência de cardiomiopatia no primeiro ano de vida e a progressão lenta da miopatia são características da LOPD.1,4 Fadiga e fraqueza muscular de cintura e membros prejudicam a participação em atividades esportivas e, até mesmo, a execução de atividades do dia a dia.1,4
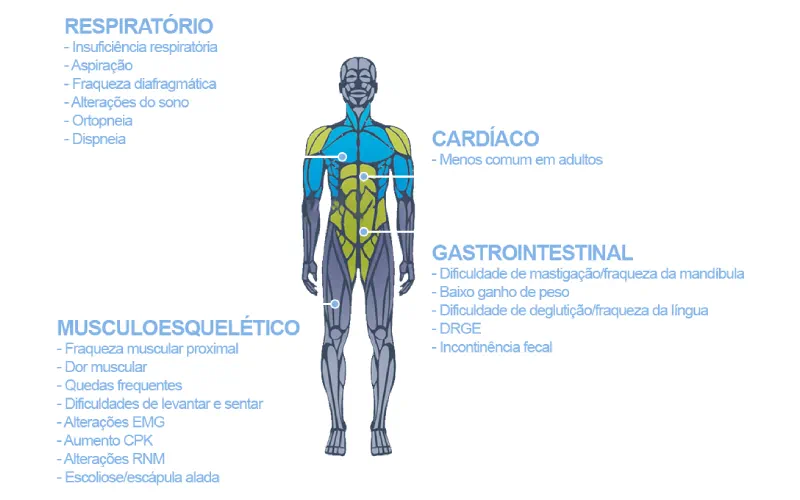
Legenda: EMG: eletromiografia; RNM: ressonância magnética; CK: creatina quinase; DRGE: doença de refluxo gastroesofágico.
Alguns sinais específicos levantam suspeitas ao longo da infância e requerem avaliação minuciosa.
Esses sinais vão desde distúrbios metabólicos até perda da fala ou marcha, entre outros sintomas neurológicos.
Para uma investigação eficiente da doença de Pompe em pacientes que apresentem sintomas característicos da doença, deve-se seguir uma estratégia laboratorial que garanta um resultado confiável no menor tempo possível. Todos os processos, desde a coleta até a análise dos resultados, devem ser realizados com cautela.

Etapa 1. Procedimento de coleta e preparo de amostra de sangue seco em papel de filtro (DBS, do inglês dried blood spot)
a) Fazer a assepsia, puncionar a ponta do dedo com uma lanceta e fazer uma leve compressão, seguida por descompressão.7 Aplicar apenas uma gota de sangue em cada círculo demarcado no papel de filtro; não é necessário preencher o círculo todo.
b) Após o preparo, deixar o DBS secando por pelo menos 4 horas em temperatura ambiente e longe de qualquer superfície absorvente.8
c) Armazenar o DBS dentro de um saco plástico com dessecante sob refrigeração até o envio para o laboratório.7 O transporte pode ser feito em temperatura ambiente.
Obs.: o material pode ser utilizado tanto para análise enzimática quanto para análise molecular.
Etapa 2. Avaliação da atividade enzimática em DBS
A determinação da atividade enzimática deve ser realizada em laboratório especializado e capacitado para tal procedimento. Os valores de referência de cada laboratório e de cada população podem variar e devem ser informados no laudo. Recomenda-se atenção ao interpretar os resultados.
Interpretação:
– Atividade enzimática compatível com DP: confirmar o diagnóstico pela pesquisa de mutações no gene GAA.9
– Atividade enzimática dentro dos valores de referência do laboratório: excluir o diagnóstico de DP.10
Etapa 3. Investigação molecular – pesquisa de mutações no gene GAA
Mais de 400 variantes patogênicas já foram descritas no gene GAA, além de inúmeras variantes consideradas benignas e outras com efeito desconhecido (VUS, de variants of unknown significance).10,11 Essas informações estão disponíveis nos bancos de dados públicos e podem ser consultadas pelos profissionais da área.
Interpretação:
– Presença de 2 mutações com efeito patogênico já descrito na literatura: diagnóstico confirmado de DP.10
– Presença de apenas 1 mutação com efeito patogênico descrito na literatura: heterozigoto para DP.10
– Presença de 1 mutação com efeito patogênico descrito na literatura + 1 mutação não descrita, de efeito desconhecido ou 1 polimorfismo: análise inconclusiva. Nessa situação deve-se avaliar a atividade enzimática em outro material biológico (pode tratar-se de heterozigoto ou de paciente com DP).10
– Presença de 2 mutações não descritas na literatura ou de efeito desconhecido: análise inconclusiva. Nessa situação deve-se avaliar a atividade enzimática em outro material biológico (pode tratar-se de heterozigoto ou de paciente com DP).10
– Ausência de mutações: caso esteja dentro da normalidade, descartar doença de Pompe. Caso esteja abaixo da normalidade, confirmar com a atividade enzimática em outro tecido.10
Etapa 4. Investigação da atividade da enzima α-glicosidase ácida em fibroblastos ou leucócitos.12,13
Para essa avaliação é necessário enviar o material específico de análise, isto é, sangue total em tubo com heparina ou fragmento de biópsia de pele para análise em leucócitos ou fibroblastos, respectivamente.8,9
Recomenda-se atenção ao interpretar os resultados, pois os valores de referência variam de acordo com o tecido estudado, a população e o laboratório em que foi feita a análise.
Interpretação:
– Atividade enzimática compatível com DP: diagnóstico confirmado de DP.9
– Atividade enzimática dentro dos valores de referência do laboratório: excluir o diagnóstico de DP.9
Doença de Pompe de início tardio – Diagnóstico diferencial
A enfermidade recebe essa classificação quando se manifesta após o primeiro ano de vida, mais comumente na fase adulta. Ocorre que seus sinais e sintomas podem se confundir com os de doenças ortopédicas, reumatológicas, cardiológicas, pulmonares e neurológicas.
Conhecer as particularidades da doença de Pompe favorece a detecção precoce, garantindo um melhor prognóstico para os pacientes. Por isso, neste vídeo, o Dr. André Macedo Serafim da Silva, neurologista e pesquisador do Hospital das Clínicas da Universidade de São Paulo (USP), esclarece o passo a passo para o diagnóstico diferencial da doença de Pompe de início tardio. Vale a pena conferir!
Doença de Pompe de início na infância – Diagnóstico diferencial
Quando a condição se manifesta ainda no primeiro ano de vida, ela é classificada como doença de Pompe de início na infância ou forma infantil da doença.
É fundamental que pediatras e neuropediatras conheçam as particularidades da enfermidade, assegurando a detecção precoce, o manejo adequado e, consequentemente, um melhor prognóstico para os pacientes. No entanto, seus sinais e sintomas, comuns a outras patologias, podem dificultar o diagnóstico. Por isso, neste vídeo, o Dr. André Macedo Serafim da Silva, neurologista e pesquisador do Hospital das Clínicas da Universidade de São Paulo (USP), esclarece o passo a passo para o diagnóstico diferencial da doença de Pompe no começo da vida.
A importância do screening familiar
Como a doença de Pompe tem origem genética e caráter hereditário, uma estratégia eficaz para identificar pacientes com essa condição é a investigação genética de seus familiares já diagnosticados.
Neste vídeo, o Dr. André Macedo Serafim da Silva, neurologista do Hospital das Clínicas da Universidade de São Paulo (USP), apresenta um caso de paciente com 11 irmãos, explicando, em detalhes, como foi realizado o screening familiar e o seu resultado: a detecção de outros oito casos da doença na família. Clique para assistir.

Historicamente, o manejo da doença de Pompe tem sido focado no controle dos sintomas, bem como em medidas gerais de suporte; e estes continuam a ser os componentes fundamentais na abordagem terapêutica geral.
Por apresentarem um amplo espectro de manifestações clínicas e diferentes tipos de incapacidade funcional, os pacientes com a doença de Pompe são melhor acompanhados por uma equipe multidisciplinar, liderada por um médico com experiência no tratamento da doença de Pompe ou doenças semelhantes,6,13 incluindo a associação de tratamento específico com medidas gerais de suporte.
RECOMENDAÇÕES PARA O MANEJO MULTIDISCIPLINAR
As doenças de depósito lisossômico, como é o caso de Pompe, são caracterizadas por manifestações multisistêmicas, necessitando médicos de diversas especialidades para realizar o acompanhamento adequado dos pacientes no monitoramento e prevenção das complicações.
Na doença de Pompe, as especialidades médicas envolvidas são cardiologistas, pediatras, clínicos gerais, ortopedistas, neurologistas, pneumologistas e outras que sejam necessárias durante o acompanhamento a longo prazo do paciente. Além do tratamento médico específico, o paciente com doença de Pompe necessita tratamento de suporte de enfermagem, fisioterapia motora e respiratória, terapia ocupacional, psicologia, fonoaudiologia e nutrição. O aconselhamento genético é fundamental para esclarecer ao paciente e a seus familiares a origem da doença e contribuir para fins de planejamento familiar.
A ENFERMAGEM E O ATENDIMENTO AOS PACIENTES.14,15
A participação da equipe de enfermagem tem início na coleta de amostras biológicas para realização de exames, que serão utilizadas para fazer o diagnóstico e dar informações gerais do estado de saúde do paciente.
O acompanhamento da terapia de reposição enzimática (TRE), preconizada para esses pacientes, começa na consulta de enfermagem, praticada por enfermeiro, na qual são orientadas as possíveis intercorrências durante a TRE e a importância da adesão ao tratamento - que uma vez iniciado não deverá ser interrompido. Quinzenalmente, devem ser verificados peso e altura. É imprescindível verificar pressão arterial, saturação, frequência cardíaca, respiração, temperatura e pulso antes, durante, a cada 30 minutos, e após a infusão.
O paciente recebe a orientação de não comparecer para realizar infusão em vigência de febre, gripe, infecções ou quaisquer outras alterações importantes. Assim, deve entrar em contato com a equipe para informar as intercorrências ocorridas para que seja dada autorização ou não para realização da infusão.
A TRE é realizada por via intravenosa e, na maioria das vezes, não apresenta dificuldade. Porém, antes de realizar a punção é necessário verificar os sinais vitais. Ressalta-se ainda que a equipe de enfermagem deve sempre estar atenta para eventuais reações adversas, pois o paciente é orientado para comunicar qualquer alteração que venha a sentir.
A relação da equipe de enfermagem com o paciente é muito estreita, pela proximidade do cuidado, frequência quinzenal e pela confiança que o paciente desenvolve pela equipe.
Quando o paciente chega ao centro de infusão ele deve relatar suas alterações de comportamento ou as manifestações clínicas ocorridas entre uma infusão e outra, sendo estes relatos registrados.
ATUAÇÃO FISIOTERÁPICA 13,15,16,17
A atuação na doença de Pompe, como em todas as doenças neuromusculares, deve ser pautada no entendimento fisiopatológico e cinesiopatológico da doença, na sua apresentação, evolução e avaliação individualizada. A chave da conduta fisioterapêutica está na compreensão da interação entre a presença, progressão e potencial remediação da fraqueza muscular; no conhecimento da biomecânica correta do movimento; no risco do desenvolvimento de contraturas e deformidades além de estratégias para sua prevenção e tratamento.
A heterogeneidade de sintomas, primariamente relacionados à disfunção progressiva da musculatura esquelética e respiratória, tem proporcionado uma variedade de intervenções fisioterapêuticas destinadas a minimizar os danos funcionais causados pela doença de Pompe. Por conseguinte, o tratamento fisioterapêutico deve ser respiratório e motor.
A assistência fisioterapêutica respiratória deve ter como objetivos principais:
- evitar a obstrução brônquica por secreções, minimizando as complicações pulmonares decorrentes de atelectasias, infecções pulmonares e disfunção diafragmática;
- promover adequada ventilação pulmonar, otimizando a distribuição da ventilação e da oxigenação;
- diminuir o trabalho ventilatório, reduzindo o consumo de energia e facilitando a ação dos músculos ventilatórios;
- evitar a intubação intratraqueal e suas complicações; auxiliar na implementação da ventilação não invasiva por pressão positiva.
A assistência fisioterapêutica motora deve, através de um programa efetivo e específico às necessidades de cada paciente, promover a prevenção de rigidez muscular, contraturas, deformidades posturais e, principalmente, prevenir e/ou minimizar o catabolismo muscular, favorecendo a síntese proteica em diferentes idades, por meio de exercícios funcionais submáximos, individualmente prescritos e monitorados. Tradicionalmente, a recomendação é que a terapia de exercícios seja de intensidade submáxima, evitando exercícios com resistência excessiva e excêntricos, devido ao risco de exacerbação da degeneração muscular.
Para a prevenção de encurtamentos musculares, contraturas e deformidades posturais são indicados exercícios de alongamento muscular, a fim de aumentar a flexibilidade do movimento e otimizar as propriedades tensão-comprimento do músculo, aumentando sua eficiência mecânica.
A fisioterapia é um importante componente no tratamento na doença de Pompe e deve ser destinada a otimizar e preservar a função fisiológica e motora, o máximo possível; minimizar o impacto clínico do processo da doença; prevenir e minimizar as complicações secundárias; promover e manter o nível máximo de função e independência funcional; maximizar os benefícios de quaisquer outras terapias que possam se tornar disponíveis, contribuindo com a melhora na qualidade de vida.
A primeira e principal equipe que deve ser formada é a do paciente e sua família que deve se unir aos profissionais da área da saúde. Essa interação é fundamental para o sucesso de todo e qualquer processo de tratamento, inclusive o fisioterapêutico.
ATENDIMENTO PSICOLÓGICO
Diante de um quadro sintomático, quando o paciente já nota as limitações impostas pela doença nas atividades cotidianas, o recebimento de um diagnóstico correto promove certo alívio. Ele passa a compreender melhor seus sintomas e demonstra certa ansiedade para iniciar o tratamento, pois muitas vezes já percorreu diversos serviços em busca de respostas. Ele, em geral, tende a aderir melhor ao tratamento e às orientações.
No paciente adulto com Pompe, pouco sintomático, o diagnóstico tem grande impacto e ele poderá levar um tempo até compreender todas as informações recebidas, podendo negar o diagnóstico e resistir (demorar) a realizar os exames. Ele demora a tomar decisões a respeito do tratamento e acompanhamento com a equipe multiprofissional. Esse tempo necessário ao paciente para aceitação do diagnóstico e da necessidade de tratamento deve ser respeitado pelos profissionais.
O acompanhamento individual com psicólogo colabora com esse processo, ajudando a esclarecer os medos e fantasias a respeito da doença crônica. O atendimento psicológico é o espaço para refletir sobre si e sobre estar doente, permitindo a expressão de sentimentos (choro e raiva) de forma acolhedora.
A doença de Pompe, como qualquer outra doença crônica, pode trazer impactos na qualidade de vida e na produtividade dos pacientes.
Do ponto de vista funcional, ela causa limitação da capacidade física, devido à diminuição da força muscular, da capacidade aeróbia e redução da resistência muscular. Essa manifestação pode, em graus variáveis de incapacidade funcional e dificuldades de mobilidade, gerar dor, diminuição da propriocepção e perda da estabilidade. Nesse aspecto, essas manifestações geram grande preocupação, fantasias, medo de ir para cadeira de rodas e de perder a capacidade de realizar as tarefas da vida diária, ou mesmo a ideia de que a mobilidade física fique prejudicada, remetendo ao sentimento de inutilidade, o que implica na diminuição da autoestima e na angústia de perder a autonomia.18-19
O paciente com a doença de Pompe geralmente não relata os sinais de depressão (como o choro constante, a falta ou excesso de apetite, a irritabilidade, entre outros) durante a consulta médica. Por outro lado, na sessão com o psicólogo, o paciente sente o acolhimento necessário e a liberdade para relatar fatos atuais da vida. Com a possibilidade de expressar os sentimentos, pode sentir-se aliviado. Caso o psicólogo perceba a persistência dos sintomas, ele deve encaminhar o paciente a um médico psiquiatra, que fará uma avaliação e o acompanhamento medicamentoso dos sintomas, se necessário. É importante salientar que o acompanhamento conjunto com o psicólogo deve ser preservado, facilitando a recuperação do paciente.
ATENDIMENTO NUTRICIONAL
O tratamento dietético com maior aporte de proteína nos pacientes com doença de Pompe foi relatado por Slonim e colaboradores21 em um menino de 7 anos de idade, com melhora na função muscular. Desde então, os relatos sobre o benefício do tratamento dietético com foco no aporte aumentado de proteínas têm sido contraditórios. No entanto, Bodamer et al.,22 em 1997, em um estudo envolvendo três pacientes, com idade entre 13-18 anos, encontraram aumento na quebra de proteína muscular, na síntese proteica e no gasto energético em repouso em relação aos controles, o que pode sinalizar uma maior demanda energética e proteica desses pacientes.
Recentemente, estudos demonstraram alterações na composição corporal, com aumento de massa gorda, no Índice de Massa Corporal (IMC), magreza ou sobrepeso, associadas à condição, bem como a diminuição da densidade mineral óssea.23-26 Em adição a estes achados, Cupler EJ, em 2012, no consenso para o tratamento da doença de Pompe com início tardio,27 chama a atenção para o aparecimento da disfagia e, consequentemente, a diminuição da ingestão de alimentos -principalmente da proteína, que pode levar ao aumento do catabolismo proteico.
Atualmente, o tratamento dietético para doença de Pompe permanece controverso, mas os estudos apontam para:
- a adequação da ingestão energética, lembrando que alguns pacientes apresentam limitações de mobilidade, que podem interferir no requerimento energético e levar ao aumento de peso, dificultando mais a locomoção e consequentemente interferindo no tratamento fisioterápico;
- a atenção à ingestão de micronutrientes, principalmente o cálcio e a vitamina D;
- e sugestão de uma dieta hiperproteica, com a ingestão de proteína em torno de 25% do total calórico.
A avaliação do estado nutricional (avaliação de peso, da composição corporal, de parâmetros bioquímicos e de consumo alimentar) e a orientação nutricional individualizada e adequada parece ser a abordagem nutricional mais apropriada para esses pacientes.
ATUAÇÃO FONOAUDIOLÓGICA
A atuação fonoaudiológica frente aos pacientes com a doença de Pompe possibilita uma nova forma de analisar, reconhecer e intervir nas funções do sistema estomatognático (sucção, mastigação e deglutição), bem como postura, tônus, mobilidade, força e sensibilidade dos músculos dos órgãos fonoarticulatórios (OFA), sendo eles maxila, mandíbula, lábios, assoalho da boca, bochechas, língua, dentes, palato duro, palato mole, arcos palatoglosso e palatofaríngeo (pilares anteriores e posteriores).
A disfunção da deglutição atribuida à fadiga dos músculos da face, mandíbula, hipotonia facial, fraqueza e alargamento da língua, dificultando o vedamento labial e dificuldades com a mastigação e deglutição (disfagia), pode ser diagnosticada com avaliação clínica e exame de videodeglutograma nas duas formas, infantil e tardia.28,29
A reabilitação fonoaudiológica para disfagia varia de acordo com a evolução da doença e, geralmente, a conduta está relacionada à gravidade das manifestações durante a alimentação.
A intervenção fonoaudiológica deve ser precoce em crianças, principalmente nas diagnosticadas antes de um ano de idade, devido à hipotonia global interferindo na sucção e deglutição, além de outros comprometimentos durante o desenvolvimento de fala e linguagem.30,31
A disfagia orofaríngea leve é caracterizada por alterações orais com necessidade de pequenas modificações na dieta, como mudança da consistência sólida (pedaços) para amassada. Neste tipo de disfagia, podem ocorrer compensações como tosse e/ou pigarros espontâneos e eficazes.
Na presença de disfagia orofaríngea moderada, o paciente pode se alimentar de consistência pastosa (liquidificada) e espessa para líquido, utilizando técnicas específicas (cabeça fletida ou lateralizada) para minimizar os riscos de aspiração e/ou facilitar a deglutição - há necessidade de supervisão de um profissional ou cuidador.
A disfagia orofaríngea grave representa impossibilidade de alimentação via oral: engasgo com todas as consistências, aspiração silente para duas ou mais consistências, tosse voluntária ineficaz e inabilidade de iniciar a deglutição. A reabilitação fonoaudiológica nos casos graves é baseada na restrição de consistências alimentares e com auxílio de manobras posturais para evitar o uso precoce de vias alimentares alternativas. Para os pacientes que conseguem manter uma alimentação via oral, são realizados exercícios para melhorar a mobilidade, o tônus e a força de OFA com menor intensidade devido à fadiga dos músculos da face, dos lábios, da língua e das bochechas.6
Os pacientes com início tardio da doença podem apresentar dificuldade respiratória e apneia do sono, além da queixa da fala (disartria) e alteração da qualidade de voz (anasalada) por incompetência velofaríngea.31
Podemos concluir que o papel do fonoaudiólogo com esses pacientes é fundamental para promoção de qualidade de vida e minimização dos riscos de aspiração alimentar, propiciando manobras terapêuticas e orientação aos familiares e cuidadores.
Devido à raridade da doença de Pompe e a manifestação de sinais e sintomas que confundem-se com os de outras doenças neuromusculares, o diagnóstico geralmente é obtido após um período muito longo, adiando a implementação de estratégias terapêuticas.32
Analisando-se estudos de caso, nota-se que os registros de atraso de diagnóstico se concentram, principalmente, em pacientes com idade avançada. Ou seja, o maior conhecimento da doença no contexto atual permite antecipar a detecção do problema nas novas gerações. Vale ressaltar que pacientes mais jovens e menos afetados respondem mais favoravelmente à terapia de reposição enzimática.32
Uma análise dos dados da história natural da doença em crianças e adultos demonstrou que, a cada ano em que a doença de Pompe permanece sem tratamento, aumenta a probabilidade de o paciente necessitar de cadeira de rodas ou de suporte respiratório.33
Outra pesquisa, publicada pelo periódico Neurology, observou a relação entre a gravidade da doença de Pompe (evidenciado pelo uso de cadeira de rodas ou suporte respiratório), a idade dos pacientes e o tempo decorrido a partir das primeiras manifestações, em um grupo de 255 crianças e adultos. A gravidade da doença não se mostrou relacionada à idade dos participantes, mas, sim, ao tempo transcorrido a partir das primeiras manifestações.
Sixel et al32 também observou uma relação direta entre a demora no diagnóstico e a ocorrência de complicações. Em 2017, um estudo com 6 pacientes apontou que a média de idade em que eles receberam o diagnóstico foi de 39, 5 anos. E o intervalo entre o início dos sintomas e a detecção da enfermidade foi de 8 anos. No momento do diagnóstico, os pacientes já apresentavam sintomas iniciais de fraqueza motora.
Diante de tais evidências, fica clara a importância de correr contra o relógio, diante do surgimento dos primeiros sintomas. Em outras palavras, um diagnóstico precoce permite um acompanhamento multidisciplinar adequado, visando retardar/estabilizar a progressão da doença e ampliando consequentemente o prognóstico favorável ao paciente.
- Neurologista
- Geneticista
- Reumatologista
- Pneumologista
1. Van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008;372:1342‐1353.
2. Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7(6):713-6.
3. Lagler FB, Moder A, Rohrbach M, Hennermann J, Mengel E, Gökce S, et al. Extent, impact, and predictors of diagnostic delay in Pompe disease: A combined survey approach to unveil the diagnostic odyssey. JIMD Rep. 2019;49(1):89-95.
4. Chan J, Desai AK, Kazi ZB, Corey K, Austin S, Hobson-Webb LD, et al. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol Genet Metab. 2017;120(3):163-172.
5. Doença de Pompe. Hospitais Federais Universitários – Ministério da Educação. Disponível em: http://www2.ebserh.gov.br/web/hu-ufjf/doenca-de-pompe. Acesso em maio de 2020.
6. Kishnani PS, Steiner RD, Bali D et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8:267-288.
7. Zhang H, Kallwass H, Young SP, Carr C, Dai J, Kishnani PS, et al. Genet Med. 2006 May;8(5):302-6
8. Müller KB, Mayra DB Rodrigues MDB, Pereira VG, Martins AM, D’Almeida V. Reference values for lysosomal enzymes activities using dried blood spots samples − a Brazilian experience. Diagn Pathol. 2010;5:65.
9. Winchester B, Bali D, Bodamer OA, Caillaud C, Christensen E, Cooper A, et al; Pompe Disease Diagnostic Working Group. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: report from an international consensus meeting. Mol Genet Metab. 2008 Mar;93(3):275-81.
10. Burton BK, Kronn DF, Hwu WL, Kishnani PS; Pompe Disease Newborn Screening Working Group. The initial evaluation of patients after positive newborn screening: recommended algorithms leading to a confirmed diagnosis of Pompe disease. Pediatrics. 2017 Jul;140(Suppl 1):S14-S23.
11. Reuser AJJ, van der Ploeg AT, Chien YH, Llerena J Jr, Abbott MA, Clemens PR, et al. GAA variants and phenotypes among 1,079 patients with Pompe disease: data from the Pompe Registry. Hum Mutat. 2019 Jul 24.
12. Chamoles NA, Niizawa G, Blanco M, Gaggioli D, Casentini C. Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin Chim Acta. 2004 Sep;347(1-2):97-102.
13. Case LE, Kishnani PS. Physical therapy management of Pompe disease. Genet Med. 2006;8(5):318-27.
14. Brunoni D, Perez, ABA. Guias de Medicina Ambulatorial e Hospitalar da EPM-UNIFESP: Genética Medica Manole, 2013. PP 709.
15. Ausems MG, Lochman P, van Diggelen OP, et al. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology 1999;52(4):851-3.
16. Bembi B, Cerini E, Danesino C, et al. Management and Treatment of Glycogenosis Type II. Neurology 2008;71:S12
17. Favejee MM, Huisstede BMA, Bussmann JBJ, Kruijshaar ME, Van der Ploeg AT. Physiotherapy Manangement in late-onset Pompe disease: Clinical practice in 88 patients. Molecular Genetics and Metabolism 2012; 107: 111-5.
18. Kayser, Bárbara, Miotto, Cascieli, Molin, Vinicius Dal, Kummer, Julia, Klein, Suelén Roberta, & Wibelinger, Lia Mara. (2014). Influência da dor crônica na capacidade funcional do idoso. Revista Dor, 15(1), 48-50.
19. Nunes, Nunes, Lorena, Novo, Juliano, Schnaider. (2014). Autoestima, depressão e espiritualidade em pacientes portadores de doença renal crônica em tratamento hemodialítico. Revista do Médico Residente, 16(1).
20. Lima, Carvalho, Silva, Gomes, Passos e Santos (2012) Diagnostico de enfermagem evidenciados em mulheres com feridas crônicas in Revista Baiana de enfermagem, vol 26 no.3 ISSN 2178-8650 (eletrônico).
21. Slonim AE, Bulone L, Goldberg T, Minikes J, Slonim E, Galanko J, et al. Modification of the natural history of adult-onset acid maltase deficiency by nutrition and exercise therapy. Muscle Nerve. 2007; 35:70-7.
22. Bodamer OAF,Leonard JV, Halliday D. Dietary treatment in late-onset acid maltase deficiency. Eur J Pediatr (1997) 156 [Suppl 1]:S39S42.
23. Ravaglia S, Pichiecchio A, Rossi M, Filippi PD, Minelli A, Moglia A, et al. Dietary treatment in adult-onset type II glycogenosis. J Inherit Metab Dis. 2006; 29:590.
24. Ravaglia S, Danesinob C, Mogliaa A, et al. Nutritional status and body composition during enzyme replacement therapy in adult-onset type II glycogenosis. European Journal of Neurology 2010, 17:957-62.
25. Papadimas GK, Terzis G, Methenitis S, et al. Body composition analysis in late-onset Pompe disease. Molecular Genetics and Metabolism 102 (2011) 41-3.
26. Terzis G, et al. Effect of aerobic and resistance exercise training on late-onset Pompe disease patients receiving enzyme replacement therapy. Mol. Genet. Metab. (2011) Nov;104(3):279-83. doi: 10.1016/j.ymgme.2011.05.013. Epub 2011 May 19.
27. Cupler EJ, Berger KI, Leshner RT, et al. Consensus treatment recommendations for late onset pompe disease.Muscle Nerve. 2012 March; 45(3): 319-33.
28. Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr. 2004;144:S35-S43.
29. AANEM Practice Topic: Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve. 2009;40:149-60.
30. Lisa D, Hobson-Webb A, Harrison N, Jones B, Kishnani PS. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscular Disorders 23 (2013) 319-23.
31. Jones HN, Muller CW, Lin M, Banugaria SG, Case LE, Li JS, OGrady G, Heller JH, Kishnani PS. Oropha-ryngeal Dysphagia in Infants and Children with Infantile Pompe Disease. Dysphagia (2010) 25:277-83.
32. Sixel B, Silva L, Cavalcanti N, et al. Respiratory Manifestations in Late-Onset Pompe Disease: A Case Series Conducted in Brazil. J Bras Pneumol. 2017;43(1):54-59.
33. Hagemans MLC, Winkel LPF, Van Doorn PA, et al. Disease Severity in Children and Adults With Pompe Disease Related to Age and Disease Duration. Neurology. 2005 Jun 28;64(12):2139-41.
34. Hirschhorn R, Reuser AJJ. Glycogen Storage Disease Type II: Acid Alphaglucosidase (Acid Maltase) Deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th Edition. New York: McGraw-Hill; 2001. p. 3389-420.
35. Schoser B, Stewart A, Kanters S, et al. J Neurol. 2017 Apr;264(4):621-630
36. Chien YH, Tsai FJ, Shieh JY. Long-term Prognosis of Patients With Infantile-Onset Pompe Disease Diagnosed by Newborn Screening and Treated Since Birth. J Pediatr. 2015 Apr;166(4):985-91.e1-2
GZBR.MYOZ.20.05.0196/Maio2020
Compartilhar