O que é a doença de Pompe?
É uma doença neuromuscular genética degenerativa, progressiva e hereditária que afeta pacientes de todas as idades.1,3,4 Ou seja, ela não é contagiosa, quem tem doença de Pompe nasce com ela. Aproximadamente 1 em 40.000 pessoas são acometidas.6
O Dia Nacional de Conscientização sobre a Doença de Pompe é lembrado em 28 de junho, quando são destacadas informações sobre essa doença rara, como a importância do diagnóstico precoce e seu impacto na qualidade de vida dos pacientes.
Fisiopatologia
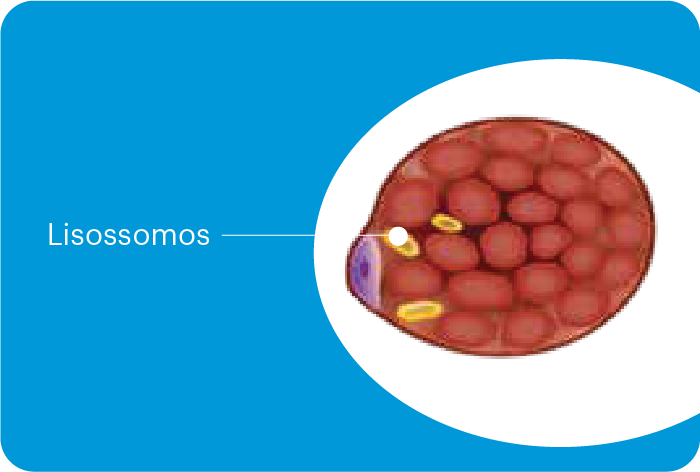
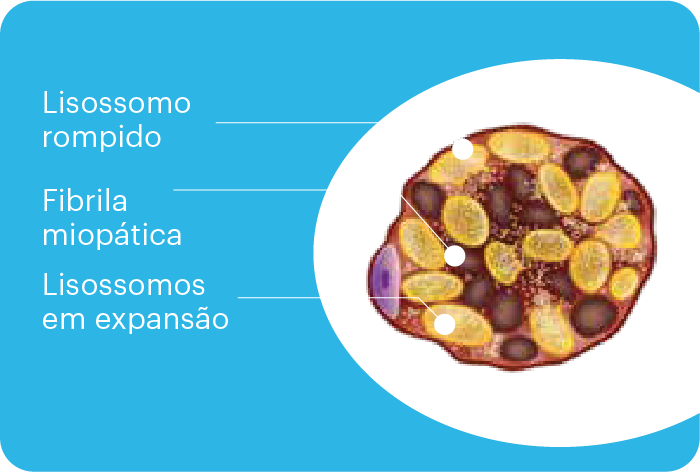
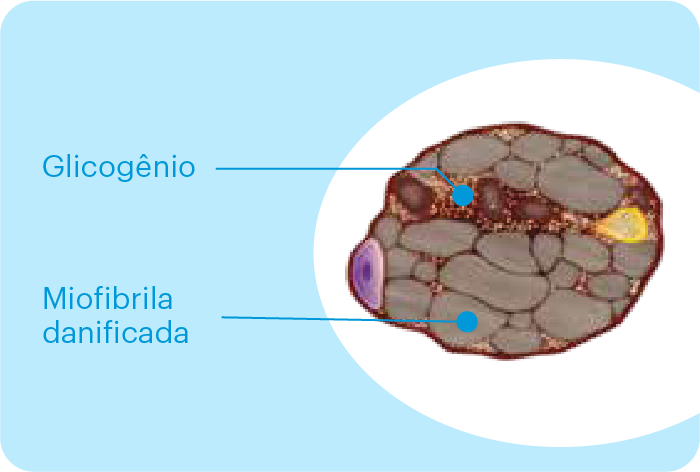
Os músculos do nosso corpo podem armazenar energia por meio de um açúcar chamado glicogênio. A doença de Pompe se manifesta quando nosso organismo não produz, ou produz em pequenas quantidades, uma enzima chamada alfa-glicosidase ácida (GAA), responsável justamente por quebrar este glicogênio e produzir energia.14,63,1
Com o tempo, esse açúcar acumulado nas células leva a danos que comprometem a musculatura do paciente, causando fraqueza muscular e respiratória.1

Como é a transmissão genética?
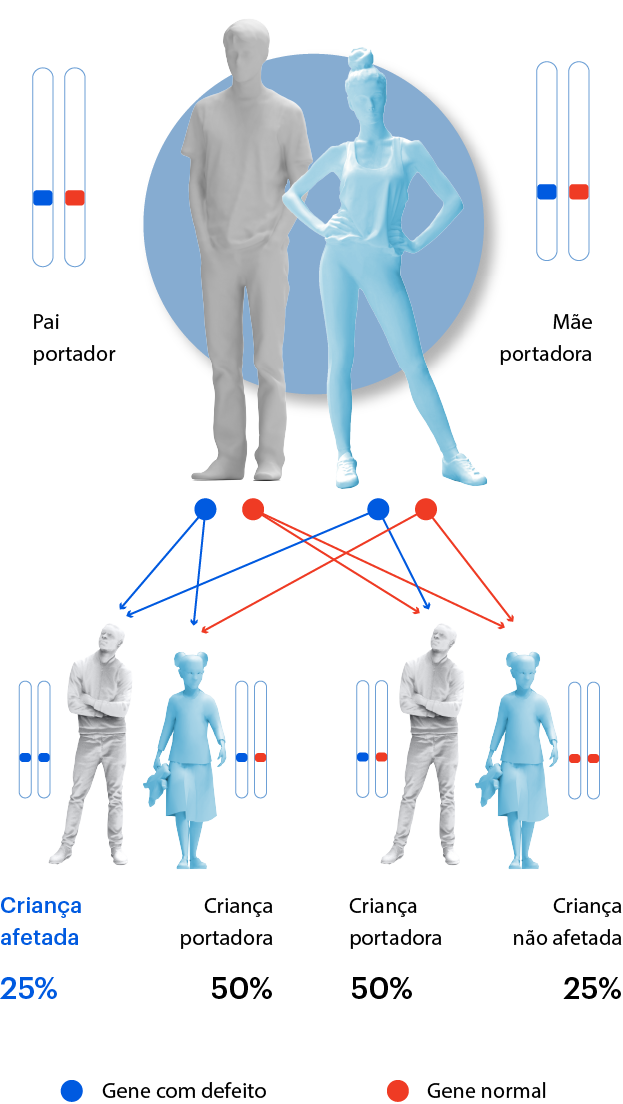
É possível carregar o gene da enzima GAA com defeito, mas não ter a doença. Quem tem apenas um dos genes defeituosos é chamado de portador e não apresenta sintomas. Já quem tem as duas cópias do gene GAA defeituoso, é afetado pela doença.
Ou seja, os portadores não têm risco de desenvolver a doença, mas podem transmitir o defeito para seus filhos. Caso ambos os pais sejam portadores, ainda assim a chance da criança nascer com Pompe é de 25%.
Se alguém na sua família já foi diagnosticado, procure um médico e faça um teste para diagnóstico.
Transmissão (árvore genealógica)
Sinais e sintomas

77% dos pacientes
com a doença de Pompe de início tardio (LOPD) apresentam intolerância a exercícios e fraqueza muscular.2

55% dos pacientes
com a doença de Pompe de início tardio (LOPD) apresentam insuficiência respiratória e fraqueza muscular.2


Dificuldade de:
- levantar os braços
- subir escadas
- levantar-se
- caminhar
Como é feito o diagnóstico?
Pacientes com LOPD podem levar de 6 a 12 anos para receber um diagnóstico deinitivo27
Para determinar se uma pessoa tem ou não a doença de Pompe, podem ser feitos diferentes exames, que incluem testes musculares, de movimento e respiratórios. Caso haja suspeita, um teste simples e específico, utilizando algumas gotas de sangue, ou um teste genético podem confirmar o diagnóstico.3
Doenças raras apresentam inúmeros desafios, como falta de informação, dificuldade e atraso no diagnóstico e ausência de opções de tratamento para muitas delas. Nesse cenário, os médicos desempenham um papel fundamental para a identificação dos sintomas e a realização do diagnóstico precoce.
Tem dúvidas e está buscando suporte médico?
-
Historicamente, o manejo da doença de Pompe é focado no controle dos sintomas, bem como em medidas gerais de suporte, e esses continuam a ser os componentes fundamentais na abordagem terapêutica geral.
Por apresentarem um amplo espectro de manifestações clínicas e diferentes tipos de incapacidade funcional, os pacientes com a doença de Pompe são melhor acompanhados por uma equipe multidisciplinar, liderada por um médico com experiência no tratamento da doença de Pompe ou doenças semelhantes,6,13 incluindo a associação de tratamento específico com medidas gerais de suporte. -
Quando possível, o atendimento multidisciplinar deve ser conduzido por um médico com experiência no tratamento da doença de Pompe ou distúrbios semelhantes, com a ajuda de especialistas apropriados.2
E quem são esses profissionais?

Neurologistas: Responsáveis pelo tratamento de doenças que afetam o sistema nervoso (cérebro, medula espinhal, raízes nervosas e nervos);
Pneumologistas: Responsáveis por tratar problemas nas vias respiratórias e pulmão;
Geneticistas: Responsáveis pelo estudo das características biológicas e sua transmissão a cada geração;
Reumatologistas: Responsáveis pelo tratamento de doenças que afetam o sistema locomotor (articulações, ligamentos, músculos, tendões e ossos);
Pediatras: Responsáveis pelo tratamento de crianças e adolescentes;
Psicólogos: Responsáveis pelo estudo da mente e comportamento humano;
Fisioterapeutas: Responsáveis por tratar doenças e lesões e recuperar movimentos físicos.
A Sanofi Genzyme desenvolveu um aplicativo com conteúdo exclusivo sobre as doenças raras. Baixe o app e saiba mais sobre as características da doença de Pompe.
Confira a vídeo aula que preparamos sobre o assunto!
-
Kishnani PS, et al;
ACMG Work Group on Management of Pompe Disease.
Genet Med. 2006;8(5):267-288. doi:10.1097/01.gim.0000218152.87434.f3. -
Schüller A, et al.
Am J Med Genet C Semin Med Genet.
2012;160C(1):80-88.doi:10.1002/ajmg.c.31322. -
Kishnani PS, et al.
J Pediatr.
2004;144(5 Suppl):S35-S43. -
American Association of Neuromuscular and Electrodiagnostic Medicine.
Muscle Nerve.
2009;40(1):149-160. doi:10.1002/mus.21393. -
van Capelle CI, et al.
Orphanet J Rare Dis.
2016;11(1):65.doi:10.1186/s13023-016-0442-y. -
Ausems MGEM, et al.
Eur J Hum Genet.
1999;7(6):713-716. doi:10.1038/sj.ejhg.5200367. -
Cupler EJ, et al;
AANEM Consensus Committee on Late-Onset Pompe Disease. Muscle Nerve.
2012;45(3):319-333. doi:10.1002/mus.22329. -
Chien YH, et al.
J Pediatr.
2011;158(6): 1024-1027.e1. doi:10.1016/j.jpeds.2010.11.053. -
Kishnani PS, et al;
Infantile-Onset Pompe Disease Natural History Study Group.
J Pediatr. 2006;148(5):671-676. doi:10.1016/j.jpeds.2005.11.033. -
Alejaldre A, et al.
Neuromuscul Disord.
2012;22(Suppl 2):S148-S154. doi:10.1016/j.nmd.2012.05.011. -
Hirschhorn R, et al.
In: Scriver CR, et al, eds. The Metabolic Bases of Inherited Disease.
8th ed. New York, NY: McGraw-Hill; 2001:3389-3420. -
Al Jasmi F, et al;
the MENA Pompe Working Group.
BMC Neurology. 2015;15:205.doi:10.1186/s12883-015-0412-3. -
Thurberg BL, et al.
Lab Invest.
2006;86(12):1208-1220. -
Raben N, et al.
Mol Genet Metab.
2010;101(4):324-331. doi:10.1016/j.ymgme.2010.08.001. -
Chan J, et al.
Mol Genet Metab.
2017;120(3):163-172. doi:10.1016/j.ymgme.2016.12.004. -
Karabul N, et al.
JIMD Rep. 2014;17:53-61.
doi:10.1007/8904_2014_334. -
Bernstein DL, et al.
Mol Genet Metab.
2010;101(2-3):130-133. doi:10.1016/j.ymgme.2010.06.003. -
Toscano A, et al.
Acta Myol.
2013;32(2):78-81. -
Preisler N, et al.
Mol Genet Metab.
2013;110(3):287-289. doi:10.1016/j.ymgme.2013.08.005. -
Manganelli F, et al.
Acta Myol.
2013;32(2):82-84. -
Moghadam-Kia S, et al.
Cleve Clin J Med.
2016;83(1):37-42.doi:10.3949/ccjm.83a.14120. -
Rigter T, et al.
Mol Genet Metab.
2012;107(3):448-455.doi:0.1016/j.ymgme.2012.09.017. -
Mellies U, et al.
Respir Med.
2009;103(4):477-484.doi:10.1016/j.rmed.2008.12.009. -
Boentert M, et al.
Int J Mol Sci.
2016;17(10):1735. doi:10.3390/ijms17101735. -
Fuller DD, et al.
Respir Physiol Neurobiol.
2013;189(2):241-249. doi:10.1016/j.resp.2013.06.007. -
Hagemans ML, et al.
Neurology.
2005;64(12):2139-2141. -
Kishnani PS, et al;
on behalf of the Pompe Registry Boards of Advisors
Am J Med Genet. 2013;161A(10):2431-2443. -
Wokke JHJ, et al.
Muscle Nerve.
2008;38(4):1236-1245. doi:10.1002/mus.21025. -
Winkel LPF, et al.
J Neurol.
2005;252(80):875-884. doi:10.1007/s00415-005-0922-9. -
Schoser B, et al.
J Neurol.
2017;264(4):621-630. doi:10.1007/s00415-016-8219-8. -
Jaradeh S.
Muscle disorders affecting oral and pharyngeal swallowing.
GI Motility Online website. https://www.nature.com/gimo/contents/pt1/full/gimo35.html. Published May 16, 2006. Accessed July 28, 2020. -
Chaudhuri A, et al.
Lancet. 2004;363(9413):978-988.
doi:10.1016/S0140-6736(04)15794-2. -
Genetics Home Reference website.
Hereditary myopathy with early respiratory failure.
https://ghr.nlm.nih.gov/condition/hereditary-myopathy-with-early-respiratory-failure. Reviewed September 2018. Accessed July 28, 2020. -
Barohn RJ, et al.
Neurol Clin.
2014;32(3):569-vii. doi:10.1016/j.ncl.2014.04.008. -
Féasson L, et al.
Ann Readapt Med Phys.
2006;49(6):289-300. doi:10.1016/j.annrmp.2006.04.015. -
Gilchrist JM.
Semin Respir Crit Care Med.
2002;23(3):191-200. doi:10.1055/s-2002-33027. -
Ozawa E, et al.
Mol Cell Biochem.
1999;190:143-151. -
Mah JK, et al.
Neuromuscul Disord.
2014;24(6):482-491.doi:10.1016/j.nmd.2014.03.008. -
Barnabei MS, et al.
Compr Physiol.
2011;1(3):1353-1363. doi:10.1002/cphy.c100062. -
Limb-girdle muscular dystrophy.
Genetics Home Reference website.
https://ghr.nlm.nih.gov/condition/limb-girdlemuscular-dystrophy. Reviewed September 2019. Accessed July 28, 2020. -
Pegoraro E, et al. In: Pagon RA, et al, eds.
GeneReviews.
Seattle, WA: University of Washington, Seattle; 1993. https://www.ncbi.nlm.nih.gov/books/NBK1408/. Published June 8, 2000. Updated August 30, 2012. Accessed July 29, 2020. -
Siciliano G, et al.
Acta Myol.
2015;34(1):3-8. -
Myasthenia gravis.
Genetics Home Reference website.
https://ghr.nlm.nih.gov/condition/myastheniagravis. Reviewed June 2016. Accessed July 29, 2020. -
Harvard Health Publishing website.
Myasthenia gravis: what is it?
https://www.health.harvard.edu/a_to_z/myasthenia-gravis-a-to-z. Published December 2018. Accessed July 29, 2020. -
Smoyer-Tomic KE, et al.
BMC Musculoskeletal Disord.
2012;13:103. doi:10.1186/1471-2474-13-103. -
National Institute of Neurological Disorders and Stroke website.
Inflammatory myopathies information page.
https://www.ninds.nih.gov/Disorders/All-Disorders/Inflammatory-Myopathies-Information-Page. Published February 27, 2017. Accessed July 29, 2020. -
Gazeley DJ, et al.
Ther Adv Musculoskeletal Disord.
2011;3(6):315-324. doi:10.1177/1759720X11415306. -
Johns Hopkins Medicine website.
Polymyositis.
https://www.hopkinsmedicine.org/health/conditions-and-diseases/polymyositis. Published February 27, 2017. Accessed July 29, 2020. -
Mastaglia FL, et al.
Rheum Dis Clin North Am.
2002;28(4):723-741. doi:10.1016/s0889-857x(02)00021-2. -
Wagner M, et al.
Neuromuscul Disord.
2013;23(1):89-92. doi:10.1016/j.nmd.2012.09.004. -
Okumiya T, et al.
Mol Genet Metab.
2006;88(1):22-28. doi:10.1016/j.ymgme.2005.10.016. -
Behjati S, et al.
Arch Dis Child Educ Pract Ed.
2013;98(6):236-238. doi:10.1136/archdischild-2013-304340. -
Bodamer OA, et al;
on behalf of the Pompe Disease Newborn Screening Working Group.
Pediatrics. 2017;140(suppl 1):S4-S13. doi:10.1542/peds.2016-0280C. -
Recommended uniform screening panel.
Health Resources and Services Administration website.
https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html. Reviewed February 2020. Accessed July 29, 2020. -
O’Callaghan C, et al.
Respirol Case Rep.
2016;4(5):e00178. doi:10.1002/rcr2.178. -
Johns MW.
Sleep.
1991;14(6):540-545. -
Wood KL.
Merck Manual Professional Version website.
https://www.merckmanuals.com/professional/pulmonary-disorders/tests-of-pulmonary-function-pft/airflow,-lung-volumes,-and-flow-volume-loop. Updated April 2020. Accessed July 29, 2020. -
Messina Z, et al.
StatPearls.
Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK551648/. Updated November 21, 2019. Accessed July 29, 2020. -
Ortiz-Prado E, et al.
Am J Blood Res.
2019;9(1):1-14. -
Barba-Romero MA, Barrot E, et al.
Rev Neurol.
2012;54(8):497-507. -
Llerena JC Jr, et al.
Arq Neuropsiquiatr.
2016;74(2):166-176. doi:10.1590/0004-282X20150194. -
van der Ploeg AT, et al.
Eur J Neurol.
2017;24(6):768-e31. doi:10.1111/ene.13285. -
Hirschhorn, R. and Reuser, A.J.J.
(2001) Glycogen Storage Disease Type II: Acid Alphaglucosidase (Acid Maltase) Deficiency.
In: Scriver, C.R., Beaudet, A.L., Sly, W.S. and Valle, M.D., Eds., The Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, New York, 3389-3420.
Referências