
- Dor neuropática e sintomas autonômicos desencadeados por danos em neurônios e axônios periféricos.36
- Comprometimento cognitivo e transtornos do humor, evidenciados por lesões de substância branca.36
- Crises episódicas (“crises de Fabry”) – dor agonizante em queimação, que se inicia nas extremidades e irradia para as partes internas dos membros e por todo o corpo.36
- Dor crônica – parestesia com queimação e formigamento.36
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
- Bernardes TP, Foresto RD, Kirsztajn GM. Fabry disease: genetics, pathology, and treatment. Rev Assoc Med Bras (1992). 2020 Jan 13;66Suppl 1(Suppl 1):s10-s16.
- Laney DA, Fernhoff PM. Diagnosis of Fabry disease via analysis of family history. J Genet Couns. 2008 Feb;17(1):79-83.
- Nowak A, Beuschlein F, Sivasubramaniam V, Kasper D, Warnock DG. Lyso-Gb3 associates with adverse long-term outcome in patients with Fabry disease. J Med Genet. 2022
- Rozenfeld, P. et al (2019). Fabry pedigree analysis: A successful program for targeted genetic approach. Molecular Genetics & Genomic Medicine, 7, e00794. https://doi.org/10.1002/mgg3 .794
- Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003 Feb 18;138(4):338-46.
- Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93(2):112-28.
- Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a "renal variant" phenotype. Kidney Int. 2003;64(3):801-7.
- Kotanko P, Kramar R, Devrnja D, Paschke E, Voigtländer T, Auinger M, et al. Results of a nationwide screening for Anderson-Fabry disease among dialysis patients. J Am Soc Nephrol. 2004;15(5):1323-9.
- Desnick RJ, et al. In: The Online Metabolic and Molecular Bases of Inherited Diseases. New York, NY: McGraw Hill; 2014. p. 1-64.
- National Kidney FoundationClinical Update Fabry Disease and Chronic Kidney Disease. 2016. Disponível em: https://www.kidney.org/sites/default/files/02-10-7244_CBG-Fabry_Bulletin-5b.pdf. Acesso em: 8 fev. 2021.
- Ramaswami U, Najafian B, Schieppati A, Mauer M, Bichet DG. Assessment of renal pathology and dysfunction in children with Fabry disease. Clin J Am Soc Nephrol. 2010;5(2):365-70
- Pisani A, Visciano B, Imbriaco M, Di Nuzzi A, Mancini A, Marchetiello C, et al. The kidney in Fabry's disease. Clin Genet. 2014 Oct;86(4):301-9.
- Najafian B, Svarstad E, Bostad L, Gubler MC, Tøndel C, Whitley C, et al. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011;79(6):663:70.
- Torra R. Renal manifestations in Fabry disease and therapeutic options. Kidney Int Suppl. 2008;(111):S29-32.
- Ortiz A, et al. Nephrol Dial Transplant. 2008;23(5):1600-7
- Schiffmann R, et al. Nephrol Dial Transplant. 2009;24(7):2102-11
- Eng CM, et al. J Inherit Metab Dis. 2007 Apr;30(2):184-92
- Terryn W, Cochat P, Froissart R, Ortiz A, Pirson Y, Poppe B, et al. Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrol Dial Transplant. 2013;28(3):505-17.
- Linthorst GE, Bouwman MG, Wijburg FA, Aerts JM, Poorthuis BJ, Hollak CE. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet. 2010;47(4):217-22.
- Monserrat L, Gimeno-Blanes JR, Marín F, Hermida-Prieto M, García-Honrubia A, Pérez I, et al. Prevalence of fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2007;50(25):2399-403.
- van der Tol L, Smid BE, Poorthuis BJ, Biegstraaten M, Deprez RH, Linthorst GE, et al. A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J Med Genet. 2014;51(1):1-9.
- Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8(9):539-48.
- Yousef Z, et al.Eur Heart J. 2013;34(11):802-8
- Patel MR, et al. J Am Coll Cardiol. 2011;57(9):1093-9.
- Kampmann C, et al. Int J Cardiol. 2008;130(3):367-73.
- Weidemann F, et al. Orphanet J Rare Dis. 2013;8:116
- Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-79.
- Fellgiebel A, Müller MJ, Ginsberg L. CNS manifestations of Fabry's disease. Lancet Neurol. 2006;5(9):791-5.
- Sims K, et al. Stroke. 2009;40(3):788-94.
- Üçeyler N, et al. Clin J Pain. 2014;30(10):915-20
- Fellgiebel A, et al. Neurology. 2009;72(1):63-8
- Moore DF, et al. Brain Res Bull. 2003;62(3):231-40
- Cole AL, et al. J Inherit Metab Dis. 2007;30(6):943-51.
- Fabry Registry. Annual Report 2010. Disponível em: www.fabry.org/fsig.nsf/PDFs/PDFsR/$File/2010_Annual_Report.pdf. Acesso em: 19 jan. 2018
- Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550-5.
- Samiy N. Ocular features of Fabry disease: diagnosis of a treatable life-threatening disorder. Surv Ophthalmol. 2008;53(4):416-23.
- C. Orssaud, J. Dufier, D. Germain. Ocular manifestations in Fabry disease: a survey of 32 hemizygous male patients. Ophthalmic Genet, 24. (2003), pp. 129-139
- T.T. Nguyen, T. Gin, K. Nicholls, et al. Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. Clin Experiment Ophthalmol, 33 (2005), pp. 164-168
- Manger B, Mengel E, Schaefer RM. Rheumatologic aspects of lysosomal storage diseases. Clin Rheumatol. 2007;26(3):335-41.
- Pisani A, Visciano B, Roux GD, Sabbatini M, Porto C, Parenti G, et al. Enzyme replacement therapy in patients with Fabry disease: state of the art and review of the literature. Mol Genet Metab. 2012 Nov;107(3):267-75.
- Fabrazyme na versão para o profissional de saúde aprovada em 27/11/2020. Disponível em: https://consultas.anvisa.gov.br/#/bulario/q/?nomeProduto=FABRAZYME. Acesso em: 08/02/2023.
Na doença de Fabry, mutações no gene GLA, localizado no cromossomo X, resultam em defeitos na síntese e/ou função da alfa-galactosidase A (alfa-gal-A).2
A alfa-gal-A é uma enzima lisossômica que metaboliza a globotriaosilceramida (também conhecida como GL-3 ou Gb-3) e assim impede seu acúmulo. A deficiência total ou parcial de alfa-gal-A leva ao depósito progressivo de glicoesfingolipídeos, sobretudo GL-3, em lisossomos de vários tipos celulares.2
Na doença de Fabry, o acúmulo de GL-3 já se inicia no período pré-natal, ou no útero, e continua ao longo da vida.3 Com o avanço da idade, o acúmulo lisossômico progressivo de GL-3 – em especial na microvasculatura – causa insuficiência renal, doenças cardíacas, AVC e morte prematura, geralmente na terceira ou quarta década de vida.11,3
Recentemente, o Liso-Gb3, principal biomarcador da atividade da doença de Fabry, também tem sido associado a fisiopatologia da doença de Fabry e aos desfechos de longo prazo da doença4
A doença de Fabry é muito variável na forma como se manifesta clinicamente. Ou seja, podemos encontrar pessoas assintomáticas a gravemente doentes1. Nessa doença progressiva, o número de órgãos afetados e sintomas apresentados pode variar de acordo com cada paciente e com a faixa etária, além de poder se manifestar de maneiras diferentes em homens e mulheres.1
Figura 1. Confira os principais sintomas da doença de Fabry por faixa etária1:
 Adultos
Adultos
.2024-04-03-14-49-02.webp)
.2024-04-03-14-49-02.webp)
.2024-04-03-14-49-02.webp)
.2024-04-03-14-49-02.webp)
.2024-04-03-14-49-02.png)
AVC em idade precoce1
 Crianças
Crianças
.2024-04-03-14-49-02.webp)
.2024-04-03-14-49-02.png)
.2024-04-03-14-49-02.png)
.2024-04-03-14-49-02.webp)

Por estar ligada ao cromossomo X, a alteração genética que causa a doença de Fabry pode ser transmitida por homens e mulheres, e as mulheres não são apenas portadoras.2
Os homens têm 100% de chance de transmitir o gene alterado às suas filhas e 0% de chance de transmiti-lo aos filhos, enquanto as mulheres com doença de Fabry têm 50% de chance de transmitir o gene alterado para cada filha ou filho, independentemente do gênero.2
Mulheres com variantes patogênicas no gene GLA podem desenvolver as manifestações clínicas de Fabry, porém devido à inativação aleatória do cromossomo X, essa manifestação pode ser bastante heterogênea entre os sistemas acometidos, apresentando de casos leves a casos tão graves quanto ao dos homens.2
Em função do padrão genético da doença, quando você identifica um paciente com Fabry, você identifica uma família com Fabry.5 A cada paciente diagnosticado, podem-se identificar, em média, de 5 a 15 familiares afetados a partir da triagem familiar.5
Figura 2: Padrão de herança da doença de Fabry1

Adaptada de: Germain DP. Orphanet J Rare Dis. 2010 Nov 22;5:30.1
A doença de Fabry não apresenta sinais e sintomas muito específicos, demonstrando semelhança com outras patologias.1 Por isso, é comum a demora no diagnóstico final.1 A doença de Fabry pode ser confundida e diagnosticada como artrite reumatoide juvenil, febre reumática, síndrome de Raynaud, neurose, lúpus, apendicite aguda ou esclerose múltipla.1,6
Figura 3 Média de atrasos no diagnóstico da doença de Fabry7:
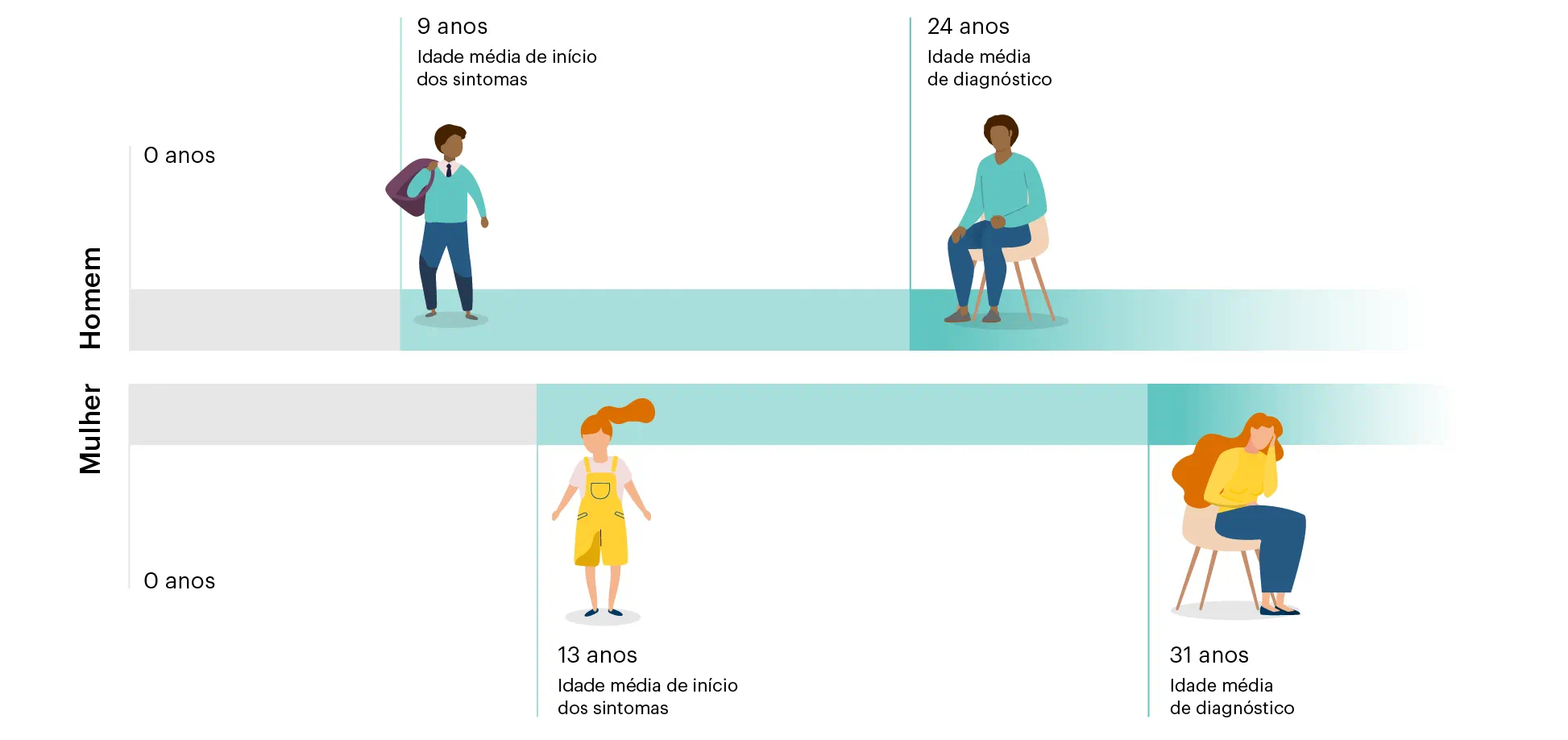
Adaptada de: Wilcox WR, et al. Mol Genet Metab. 2008;93(2):112-28.7
Embora seja considerada uma doença rara, a prevalência da doença de Fabry em pacientes com certos distúrbios, como doença renal crônica (DRC) sem causa definida, Cardiomiopatia hipertrófica (CMH) e Acidente Vascular Cerebral (AV) precoce, é superior à da população em geral.8,9 Consequentemente, em pacientes com essas manifestações clínicas, a doença de Fabry deve ser considerada um diagnóstico diferencial e os pacientes devem ser investigados8,9.
O diagnóstico em homens é feito pela atividade enzimática de alfa-galactosidase A (α-GAL) em plasma, leucócitos, fibroblastos de pele cultivados, tecido de biópsia ou sangue seco e em caso de atividade enzimática baixa, é realizada a análise de DNA para identificação da variante genética; enquanto o diagnóstico em mulheres é feito pela análise de DNA para identificação da variante genética.10
Como as mulheres com doença de Fabry podem ter atividade α-GAL em níveis baixos ou normais, deve ser realizada a análise de DNA.10 Embora mulheres com mutação no gene GLA possam ser assintomáticas ou apresentar manifestações clínicas leves, elas podem apresentar quadros tão graves quantos os homens, sendo importante o seu diagnóstico. O diagnóstico permite que profissionais monitorem sintomas novos e progressão da doença, além de ajudar a identificar outros membros da família com a doença10.
O biomarcador Liso-Gb3 somado à medição da atividade enzimática pode ser uma ferramenta de diagnóstico.6 Além disso, os níveis de Liso-Gb3 no corpo são um importante marcador da gravidade e progressão da doença, sendo um exame bastante útil no monitoramento e estadiamento da doença, inclusive para determinar o início e avaliar a resposta ao tratamento.6
 Nefrologia
Nefrologia
A doença renal é uma das principais complicações da doença de Fabry.1,11 A prevalência da doença de Fabry na população em diálise é aproximadamente 100 a 1.000 vezes superior à da população geral.12 Pacientes com doença de Fabry estão em alto risco de progredir para doença renal em estágio terminal (DRT) precocemente (30 a 50 anos e na adolescência).13
O dano renal, como resultado do acúmulo de GL-3 em várias células renais, pode se iniciar já na primeira década de vida e, geralmente, precede alterações laboratoriais e o aparecimento de sintomas clínicos14,15.
Em homens e mulheres, o acúmulo progressivo de GL-3 nos podócitos, seguido de lesão podocitária, manifesta-se posteriormente como proteinúria e redução na taxa de filtração glomerular, o que pode levar à DRC e/ou progredir para DRT14,15
Figura 4: Manifestações renais ao longo do tempo14,18

Adaptada de: 14. Najafian B, et al. Kidney Int. 2011;79(6):663:70.15 Torra R. Kidney Int Suppl. 2008;(111):S29-32. 16. Ortiz A, et al. Nephrol Dial Transplant. 2008;23(5):1600-7.17. Schiffmann R, et al. Nephrol Dial Transplant. 2009;24(7):2102-11.18. Eng CM, et al. J Inherit Metab Dis. 2007 Apr;30(2):184-92.
A European Renal Best Practice recomenda a triagem de homens abaixo dos 50 anos de idade com DRC sem causa definida e de mulheres de qualquer idade com DRC sem causa definida e outros sintomas associados à doença de Fabry19.
 Cardiologia
Cardiologia
Doença cardíaca é a causa de morte mais comum em pacientes com doença de Fabry.20,22 Manifestações cardíacas sem explicação podem indicar doença de Fabry.20,22. Estima-se que a prevalência da doença em pacientes com hipertrofia ventricular esquerda (HVE) ou CMH seja de pelo menos 1 em 100..20,22
49% dos homens e 35% das mulheres com a doença tiveram evento cardíaco com idade média de 36 e 44 anos, respectivamente17. Eventos cardíacos podem ocorrer na adolescência também.17
Hipertrofia cardíaca, fibrose e alterações de condução podem ser causadas pelo acúmulo de GL-3 nas células cardíacas, o que afeta adversamente a estrutura e a função do coração.23 Outras manifestações cardíacas podem incluir: alterações no eletrocardiograma, intervalos curtos de repolarização precoce, bloqueio atrioventricular, alterações na repolarização, alterações em ST-T e/ou arritmias.23 O acúmulo progressivo de GL-3 nos cardiomiócitos ao longo do tempo pode levar à hipertrofia ventricular esquerda (HVE) e à insuficiência cardíaca.23
Figura 5:Manifestações cardíacas ao longo do tempo17,18,24,27

Adaptada de: 17. Schiffmann R, et al. Nephrol Dial Transplant. 2009;24(7):2102-11.18. Eng CM, et al. J Inherit Metab Dis. 2007 Apr;30(2):184-92.24. Yousef Z, et al.Eur Heart J. 2013;34(11):802-8. 25. Patel MR, et al. J Am Coll Cardiol. 2011;57(9):1093-9.26. Kampmann C, et al. Int J Cardiol. 2008;130(3):367-73.27. Weidemann F, et al. Orphanet J Rare Dis. 2013;8:116.
As diretrizes da Sociedade Europeia de Cardiologia recomendam uma busca sistemática da causa subjacente de aumento da espessura na parede do VE que inclua testes laboratoriais especializados e análises genéticas.28 Dessa forma, a triagem de pacientes com HVE ou CMH sem causa definida para doença de Fabry é chave para identificar pacientes e familiares.28
 Neurologia
Neurologia
As estimativas de prevalência da doença de Fabry em pacientes com AVC precoce variam de 1% a quase 5%, muito acima da população geral.29 Com base em dados de registro, a idade média do primeiro AVC é de 39 anos em homens e 46 anos em mulheres.30
Na doença de Fabry, GL-3 pode se acumular nos neurônios periféricos e centrais, bem como nos vasos sanguíneos cerebrais, causando: 36
O acúmulo de GL-3 causa dano neural, principalmente às pequenas fibras nervosas dos sistemas nervosos periféricos somático e autônomo.36 A dor é um dos primeiros sintomas de Fabry, atingindo de 60% a 80% dos meninos e meninas afetados pelo fenótipo clássico, o que impacta a qualidade de vida.1 Os meninos geralmente são mais novos do que as meninas quando aparecem os primeiros sintomas.36 Dois tipos de dor foram descritos:1
Complicações no sistema nervoso central
As características neuropáticas periféricas iniciais da doença de Fabry são frequentemente seguidas, na idade adulta, de complicações cerebrovasculares e disfunção autonômica.29
A doença de Fabry é, muitas vezes, diagnosticada erroneamente como esclerose múltipla por conta das lesões de substância branca.29
Algumas das características neurológicas mais prejudiciais da doença de Fabry são causadas por lesões cerebrovasculares, que são resultado do envolvimento multifocal de pequenos vasos sanguíneos.30 O envolvimento cerebrovascular pode levar a uma variedade de sinais e sintomas que vão de leve a grave.30 Entre eles, estão dor de cabeça, vertigem/tontura, ataques isquêmicos transitórios e derrames isquêmicos.30
Figura 6: Manifestações neurológicas ao longo do tempo1,18,30,34

Adaptada de: 1.Germain DP. Orphanet J Rare Dis. 2010;5:30.18. Eng CM, et al. J Inherit Metab Dis. 2007 Apr;30(2):184-92.30. Sims K, et al. Stroke. 2009;40(3):788-94.31. Üçeyler N, et al. Clin J Pain. 2014;30(10):915-20.32. Fellgiebel A, et al. Lancet Neurol. 2006;5(9):791-5 .33 Moore DF, et al. Brain Res Bull. 2003;62(3):231-40.34 Cole AL, et al. J Inherit Metab Dis. 2007;30(6):943-51.
 Pediatria
Pediatria
O acúmulo de GL-3 pode se iniciar já no pré-natal ou no útero e continuar ao longo da vida.6 O início clínico da doença de Fabry ocorre na infância e sua apresentação pode ser sutil.6 Os sinais e sintomas são frequentemente interpretados como falsos ou atribuídos erroneamente a outros distúrbios, como febre reumática, eritromelalgia, neurose, síndrome de Raynaud, esclerose múltipla, polineuropatia desmielinizante crônica intermitente, lúpus, apendicite aguda, “dores de crescimento” ou petéquias.6
O curso clínico precoce da doença de Fabry costuma envolver sinais e sintomas que afetam principalmente a qualidade de vida: dor crônica, angioqueratomas, hipoidrose, intolerância a calor e a frio, e sintomas gastrointestinais.6,35
A dor é um dos primeiros sintomas de Fabry, atinge de 60% a 80% dos meninos e meninas afetados, o que reflete diretamente na qualidade de vida.1 Os meninos geralmente apresentam sintomas antes das meninas.1
Em um estudo pediátrico baseado no Registro de Fabry, meninos e meninas com a doença começam a desenvolver sintomas desde tenra idade [média de 6 anos para meninos (n=194) e 9 anos para meninas (n=158)].36 Ademais, alguns pacientes podem apresentar complicações graves precoces.36

Adaptada de:1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
 Oftalmologia
Oftalmologia
GL-3 e outros lipídeos podem se acumular nos olhos de pacientes com doença de Fabry, resultando em uma ampla gama de achados oculares e/ou queixas visuais.6,37 Esses achados não costumam afetar a visão do paciente, mas podem ser patogenomônicos para doença de Fabry.6,37
Um dos primeiros e mais comuns sinais da doença de Fabry é a cornea verticillata, observada em 53% 38 a 94% 39 dos pacientes com Fabry. O relato mais precoce em meninos é de 2 anos de idade e em meninas, de 6 anos de idade.6,37 A cornea verticillata pode ser detectada com um exame de biomicroscopia de rotina.6,37
Figura 8 : Córnea veticillata na doença de Fabry1

Adaptada de: 1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30
A cornea verticillata é um padrão simétrico, bilateral, semelhante a uma espiral, de depósitos epiteliais de GL-3 brancos, amarelos, marrons ou em pó na glândula lacrimal, emanados de um único vórtice6. Vários medicamentos, como amiodarona, também podem causar esse fenômeno e devem ser descartados.37
Além de opacidades na córnea, os pacientes com doença de Fabry podem apresentar:
• Catarata subcapsular posterior com depósitos esbranquiçados de material granular (catarata de Fabry);37
• Dilatação aneurismática em vênulas de paredes finas na conjuntiva bulbar;37
• Tortuosidade e angulação, leves a expressivas, dos vasos da retina.37
 Dermatologia
Dermatologia
A característica clínica mais marcante da doença de Fabry são os angioqueratomas, que se apresentam como:6,10
• Lesões de coloração vermelho-escura ou púrpura na pele;6,10
• Tamanho variado, de um ponto a vários milímetros de diâmetro;6,10
• Não empalidecem com pressão;6,10
• Geralmente distribuídos pelas nádegas, virilha, umbigo e coxas (distribuição de “calção de banho”).6,10
Figura 9: Angioqueratomas na doença de Fabry1

Os angioqueratomas são pequenas manchas vermelho-escuras que aumentam em número e tamanho com a idade e podem ocorrer isoladamente ou em aglomerados. Elas são normalmente encontradas na parte inferior das costas (A), nádegas (C), virilha, flancos (D) e parte superior das coxas, mas sua distribuição pode ser restrito a uma área limitada, como o umbigo (B).
Adaptada de: 1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30
As angiectasias de coloração vermelho-escura a preto-azulada são tipicamente encontradas nas regiões da coxa (à esquerda) e umbigo (à direita) (“distribuição de calção de banho”).6
As lesões geralmente aparecem durante a adolescência ou no início da idade adulta e podem se tornar maiores e mais numerosas com a idade.6 Os angioqueratomas são quase universais nos hemizigotos masculinos; aproximadamente 30% das mulheres heterozigotas apresentam alguma lesão cutânea.10
 Gastroenterologia
Gastroenterologia
Os sintomas gastrointestinais (GI) da doença de Fabry podem estar relacionados à deposição de GL-3 nos gânglios autonômicos dos intestinos e vasos sanguíneos mesentéricos.2
Apesar de comuns, os sintomas gastrointestinais ainda são uma manifestação pouco reconhecida de Fabry.1 Até dois terços dos homens afetados e cerca de metade das mulheres sintomáticas podem apresentar sintomas gastrointestinais associados à doença de Fabry.1 Os sintomas gastrointestinais costumam surgir na infância e geralmente permanecem presentes durante a vida adulta.1
Uma manifestação da doença de Fabry frequentemente negligenciada é a presença de sintomas gastrointestinais, incluindo dor abdominal (normalmente após as refeições), diarreia, náusea e vômito, que são causas relevantes de anorexia.1 A síndrome do intestino irritável (SII), com diarreia predominante, é um diferencial no diagnóstico.1
 Reumatologia
Reumatologia
Fadiga e dor podem mascarar-se como condições reumatológicas como lúpus, síndrome da fadiga crônica e artrite reumatoide ou juvenil.1,6,10,40 Em um ambiente reumatológico, os sintomas mais comuns da doença de Fabry são1,6,10,40:
• Dor nas articulações; 1,6,10,40
• Dor aguda e crônica nas mãos ou pés, ou episódios agudos de dor agonizante irradiada nas extremidades, com duração de minutos a dias (“crises de Fabry”);1,6,10,40
• Angioqueratomas: lesões cutâneas roxo-avermelhadas escuras que não embranquecem com a pressão e podem ser confundidas com vasculite;1,6,10,40
• Febre recorrente que acompanha dor e associada a taxa elevada de sedimentação de eritrócitos.1,6,10,40
A Doença de Fabry não tem cura, mas apesar de grave, tem tratamento. E quando iniciado precocemente possibilita uma melhor qualidade
de vida.1
Atualmente, o tratamento mais recomendado é a TRE (terapia de reposição enzimática). Ela é feita por meio de uma infusão sanguínea e o objetivo é repor a falta completa ou parcial da enzima, independente de qual mutação levou à doença de Fabry.2,42,42
O tratamento engloba uma abordagem multidisciplinar1,6 que pode incluir:
 Geneticista;1,6
Geneticista;1,6
Nefrologista;1,6
Neurologista;1,6
Cardiologista;1,6
Outros profissionais de saúde1,6
É importante que cada especialista monitore os pacientes com doença de Fabry quanto à emergência de novos sintomas, bem como a triagem dos sintomas existentes.1

Conheça o Programa de Suporte ao Diagnóstico e Monitoramento da Sanofi (Programa RARE) e saiba como solicitar exames para o diagnóstico diferencial para doenças raras.
Compartilhar
